
沙特医疗器械SFDA(MDMA)认证对产品有什么要求?
类别:行业资讯
文章出处:CTC华商检测
发布时间:2024-11-04 15:50:28
浏览人次:0


沙特食品药品监督管理局(SFDA) 负责监管沙特阿拉伯的医疗器械产品, 确保其安全、有效和质量稳定。所有医疗器械产品在销售产品之前必须获得SFDA批准的医疗器械上市授权medical devices marketing authorization (MDMA)。产品批准后,SFDA负责产品安全和整个生命周期管理,如MDMA变更、续证、上市后监督活动等。
SFDA医疗器械分类:
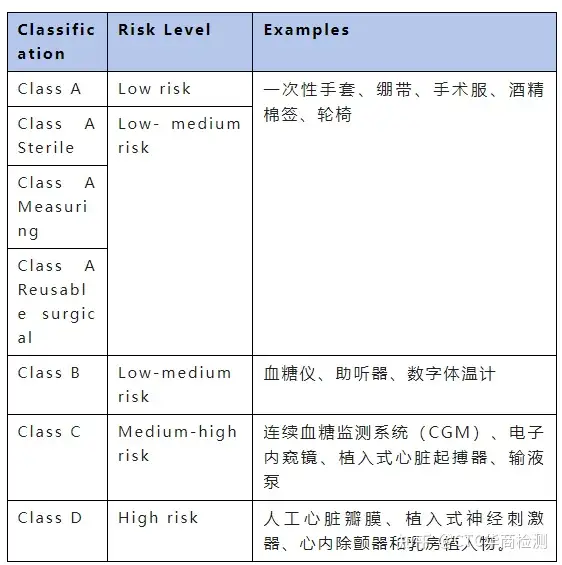
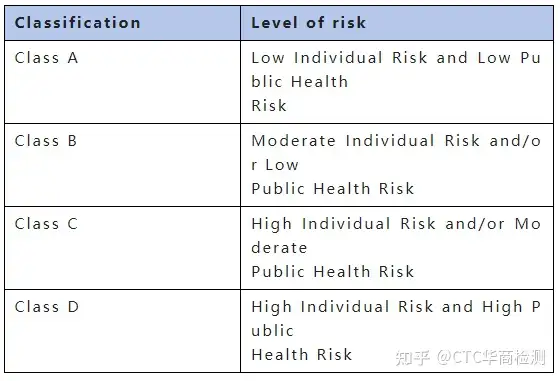
IVD医疗器械的分类:
SFDA医疗器械审批流程:
SFDA医疗器械审批包括以下五个步骤:
- 第 1 步:制造商必须根据 SFDA 的规则确定其设备的分类。
- 第 2 步:在此步骤中必须指定 KSA 授权代表 (AR)。AR 管理设备注册并代表制造商与 SFDA 进行交互。此外,KSA SR 必须向 SFDA 提交经认证的 AAR 合同以供审查。KSA SR 还将获得 SFDA 的许可,在 KSA 代表制造商。
- 步骤3:制造商将准备技术文件和申请表提交给SFDA。
- 第四步:生产企业缴纳申请费,送SFDA审核。在这里,SFDA 还可以要求提供任何其他信息。
- 第 5 步:一旦 SFDA 批准,制造商将获得其设备的医疗器械营销授权 (MDMA) 证书。
SFDA 器械审批流程所需文件
在申请设备批准时,SFDA 要求提供以下必要的文件。提交的信息必须为英文,并应包含以下内容:
设备信息:
- 商品名称为英文(如果供非专业人士使用,则为阿拉伯文)。
- 型号名称、编号和目录
- 设备描述
- 预期目的
- 分类,以及
- 其他国家/地区的批准证据(如果有)。
标签信息
- 标签和包装
- 电源标签(如适用)
- 使用说明 (IFU) 或理由信
- 有关储存、维护、运输、安装和处置的信息。
- 拟议的广告材料
有关条形码的标签信息是:
- 仅非专业用户需要它们,并且
- 有关 SFDA 对所有器械实施 UDI 要求的信息,请参阅器械唯一标识 (UDI) 部分。
在沙特阿拉伯销售医疗器械的外国制造商必须指定一名授权代表 (AR),负责向当地监管机构 - 沙特食品和药物管理局 (SFDA) 提交注册程序所需的文件,并负责上市后监督。
AR通过GHAD电子系统向SFDA转发包含产品和组织信息的医疗器械技术文件,包括预期用途、安全性和有效性、生产过程和质量控制。技术文件必须符合 MDS-REQ1 指南。SFDA 对临床和技术数据进行具体评估活动,如果结果积极,则颁发医疗器械上市许可 (MDMA)。
申请人
需要指定授权代表 (AR)。AR 必须获得 SFDA 颁发的设立许可证,并且对于 C 类和 D 类器械,提供已根据沙特标准 SFDA.MD/GSO ISO 13485:2016 或同等文件实施质量管理体系的证据。
授权代表(AR)的义务和责任:
1. 授权代表应代表制造商与SFDA打交道。
2. 授权代表应配合SFDA开展上市后监管活动。
3. 授权代表应向SFDA通报在沙特境外发生的,对在沙特境内流通的医疗器械有影响的任何事件。授权代表应说明情况并提供制造商已采取或拟采取的纠正措施的信息。
4. 授权代表应通知SFDA由制造商开展的关于沙特境内的医疗器械的任何调查所产生的所有纠正措施。授权代表应解释采取纠正措施的原因,并提供由制造商已采取或打算采取的措施的信息。
5. 授权代表应根据《医疗器械法》及其条例的规定,与从事有关在沙特流通的医疗器械的活动者进行合作。这应记录在授权代表和制造商之间的协议中。
6. 在协议中规定的授权代表对医疗器械的责任不因授权代表要求终止协议而终止,除非制造商指定其他授权代表替代,或者该医疗器械不再在市场上进行流通或者使用。
7. 当有必要终止协议时,授权代表应书面通知制造商和SFDA。
授权代表应提供SFDA要求的任何信息或相关文件。


部分过审案例





最新资讯文章
- · 沙特医疗器械SFDA(MDMA)认证对产品有什么要求?
- · 加拿大医疗器械许可MDEL注册哪里可以办理?一定要提供MDL证书吗?
- · 珠宝饰品上亚马逊/TEMU要求ASTM F2923-20怎么收费?费用和什么有关系?
- · 如何查询产品做METI备案还是结构分析函?
- · 亚马逊含纽扣电池产品或硬币电池UL4200a测试对产品电池仓结构有什么要求?
- · 什么是欧盟包装指令?包装指令在欧洲是强制的吗?
- · Grs认证有效期多久?主要审核企业哪些要求?
- · 可回收包装袋警示语应该打哪些标识?去哪里可以查询?
- · 西班牙包装回收SCRAP注册是什么?强制性要求吗?
- · 法国EPR合规包装法注册,Triman回收标识有什么要求?





